Q: I learned that initial values for differential equations need to be in ONE sequence statement. Here each is in a separate one. Which way is right?
A: If the order is important, then using one sequence statement will make sure the actions taking are in the order you wrote it.For example
sequence{A1=100
C=A1/50}
If the order is not important, as was the case in our exercise, then you can use one or more sequence statements.
For example:
sequence{A1=100}
sequence{A2=50}
Q: Can you please explain the sequence statement and its applicability again?
A: When PML code is executed the code is read at each integrated time of your numerical integration procedure. However, there are situations when you want a specific part of the code to be read and executed at specific points in time. In our case, we wanted to initialize the compartments at time=0. Here you can simply use a sequence statement like we did in our code.You can also execute a code at a later point in time. For this you can use the sleep() statement. This will tell the program to wait for a specific time before executing the next line of code. If you want to initialize Circ compartment after 10 hours, assuming the time unit is in hours, and re-initialize after 24 hours, here is the corresponding PML code:
sequence {
sleep(10)
Prol=Circ0
Sleep(14)
Circ=Circ0
}
Q: Why did you choose a saturating PK model?
A: We have used information from a literature reference to develop this model. Therein, a capacity-limited elimination had been described for the PK of the compound. Looking at the PK data, which has been digitized from the hardcopy, we might not have chosen a saturating model and a regular 1-compartment IV Bolus would have sufficed. But in order to be as close as possible to the published model we have used the Michaelis-Menten equation to describe elimination of the compound.
In general, you should be careful to fit models with saturation if the data does not support the saturation process. When doing that, you can easily see Vmax and Km being confounded. In order to resolve that issue, you need to fix one of the two parameters based on prior knowledge.
Q: How do you obtain initial estimates for Vmax and Km?
A: Km must be in the bulk of your concentration data. Therefore a good idea is just to take as initial value the average concentration level.
For Vmax you can look at the lowest and highest dose. An approximation is that at low dose C<Km and therefore you have the saturation process being linear:
Vmax*C/(C+Km)~(Vmax/Km)*C
And therefore VM/KM is in clearance units and can be estimated with NCA of the data at low doses.
Since Km is approximated already, you get an approximation for Vmax. Now you can use the Initial Estimates tab in Phoenix to fine tune that.
At high dose you converge to a zero order process:
Vmax*C/(C+Km)~Vmax
and you can test that high doses do not depend too much on Km with the Initial Estimates tab (up to a certain extent).
Q: Some versions of the Friberg Model have a mixture model for the "baseline" neutrophil count. Is there any information about when the mixture modeling will be available in Phoenix?
A: Mixture modeling is something we are considering for a future version of Phoenix NLME. Another option is to use the non-parametric method which will help you identify the observed distribution without any assumptions of normality.
Q: What factors help to choose number of compartments?
A: Typically, you are looking at diagnostics in the Overall worksheet to choose the right number of compartments. When -2LL decreases by 10 when going from 1 compartment to 2 compartments is a significant change that lets you favor the latter over the former. Same when going from 2 to 3, since these models are nested.
Q: Can the models be compared with Model comparer as well?
A: Model Comparer can only be used for analysis of population models which requires a subject identifier to differentiate data between subjects. Data for this model has been digitized from plots that is why no subject identifier was available during model building. That is also the reason why the model was built in non-population mode.
Q: How are you concluding that the 3 transit comp shows best fit?? Given that all 3 models show good fit and low errors on parameters and not much change in -2LL and AIC; whould you not choose the 2 transit comp (going with parsimony)??
A: You are absolutely correct. In a perfect scenario the model with 2 transit compartments would have been chosen, since it is the simplest model and differences in all diagnostics were non-significant. Reliance on the model with 3 transit compartments was simply based on trying to be as close as possible to the literature reference where 3 transit compartments had been proposed.
Q: slide 6: Edrug = slope x C. Slope of what?
A: The relationship between the effect and the drug concentration was assumed to be linear, that means the effect will increase proportional to the concentration. This relationship is typically described by a linear equation in the most general form: y= a*x + b. In our case, y is the effect, a is the slope, x is the concentration and there is no intercept (  .
.
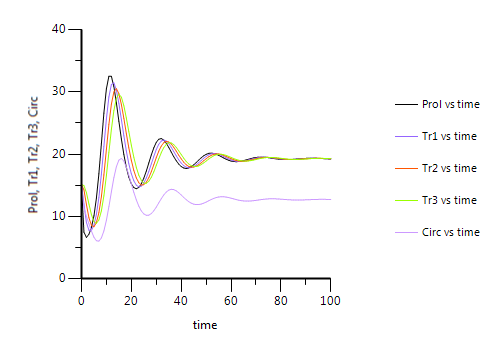
Q: In simulation plot to 100 h, Circ retuns to baseline, Prolif does not. Does this mean drug has permanent effect?
A: No, initiation of all compartments to baseline level of circulating cells was just an assumption. As we can see in the picture below, eventually all compartments will reach steady state or baseline level. It just turns out that with the final estimates of all rate constants that result from fitting of the model to the data, the production of cells in the proliferating compartment and their transport through the transit compartments is slower than the non-drug related elimination of cells from the circulating compartment.That is the reason why at steady state the proliferation and transit compartments have a higher baseline value than the circulating leukocytes.