Dear Serge / Samer,
Thank you for reply,
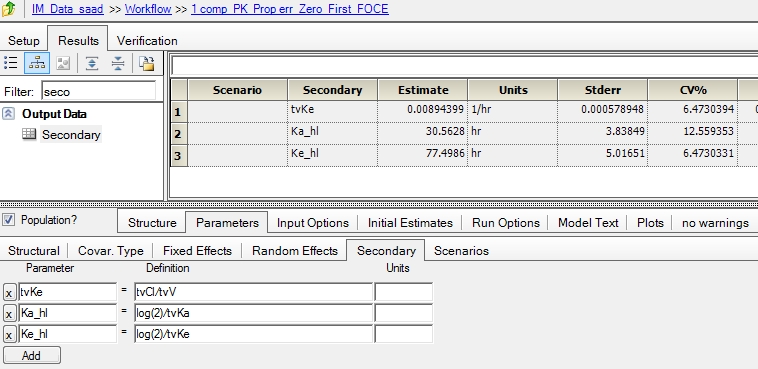
Modeled the IM data with one compartment parallel zero order and first order absorption with the following code that is mostly obtained from "Edit graphical" option.
The objective is to model the release of the drug from the IM site and also model the entry of drug in to the systemic circulation. the dose is 40 mg and so i have considered two dose points (both equal 40 mg) and parameter F is introduced. Please find the project attached for further details. The code is as follows:
test(){
deriv(A1 = - (Cl * C) + (Aa * Ka))
urinecpt(A0 = (Cl * C))
deriv(Aa = - (Aa * Ka))
C = A1 / V
dosepoint(A1, bioavail = (F), duration = (tk0), idosevar = A1Dose, infdosevar = A1InfDose, infratevar = A1InfRate)
dosepoint(Aa, bioavail = (1-F), idosevar = AaDose, infdosevar = AaInfDose, infratevar = AaInfRate)
error(CEps = 0.342651)
observe(CObs = C * (1 + CEps))
stparm(V = tvV * exp(nV))
stparm(Cl = tvCl * exp(nCl))
stparm(Ka = tvKa * exp(nKa))
stparm(F = tvF * exp(nF))
stparm(tk0 = tvtk0 * exp(ntk0))
fixef(tvV = c(, 8.37698, ))
fixef(tvCl = c(, 0.073933, ))
fixef(tvKa = c(, 0.0227794, ))
fixef(tvF = c(, 0.686753, ))
fixef(tvtk0 = c(, 303.547, ))
ranef(diag(nV, nKa, nCl, nF, ntk0) = c(0.064958412, 0.54685374, 0.30482181, 0.078260499, 0.60039497))
}
The other question is, how to simulate the steady state data for this model ???
Thanks in advance
Regards
RC
 IM_Data.phxproj 2.33MB
379 downloads
IM_Data.phxproj 2.33MB
379 downloads