Here are the materials from this session.
Slide deck:
 PK43_multiple_absoprtion_routes.pdf 375.67KB
1563 downloads
PK43_multiple_absoprtion_routes.pdf 375.67KB
1563 downloadsTextual model:
 PK43_Multiple_Absorption_Routes.txt 1.23KB
1073 downloads
PK43_Multiple_Absorption_Routes.txt 1.23KB
1073 downloadsLink to recorded webinar:
https://certara.webe...6b6aba136d78e10
The responses to questions that were raised during the Q&A session are below:
Can you explain a little bit about all the plots that Phoenix spits out?
The Phoenix model object was designed to support both individual (WNL model) and Population PK/PD modeling (NLME mode), with standard output plots that have nomenclature similar to NONMEM (DV = dependent variable, IVAR = independent variable, TAD = Time After Dose, IPRED = individual predicted, PPRED = population predicted, and so on.) Similar names are used for the output plots when models are run in Individual mode. For a glossary description of the output plots for the Phoenix Model Object, see Help | Contents | Phoenix Model | Phoenix Model Run Modes, Engines, and Output | Phoenix Model Output | Plot output.
Will there still be a session for nonlinear kinetics?
Nonlinear kinetics has already been introduced in the following PML school sessions:
Lesson 1: Nonlinear Clearance
Lesson 2: Parent and Metabolite Kinetics
Nonlinear kinetics will also be covered extensively in future webinars in the series.
Can we model (i) discontinuous absorption in Phoenix how different is it from this model? (ii) Can we build transit compartments in the absorption phase?
Yes, discontinuous absorption can be modeled using “sequence” and “sleep” statements in a PML textual model. It is also possible to build transit compartments anywhere in a Phoenix text model. The syntax for these statements are described in Help | Documents | Phoenix Modeling Language Reference Guide.pdf.
How do you choose the error model?
Dan Weiner discussed this during the Q&A session of the webinar. Typically it is a good idea to start with the default Additive error model and work on model specification and initial error estimates. Once the structural model is finalized, the model can be fine-tuned by specifying the error model. The residual plots such as Ind IWRES vs IPRED, Ind IWRES vs IVAR, and QQIWRES can be used to identify patterns that might suggest a different error model is appropriate.
Why linear up and logdown calc method was used for NCA?
The linear up log down AUC calculation method uses the linear trapezoidal rule when concentrations are increasing, and the log method when concentrations are decreasing. Linear Up Log Down is a good choice when there are multiple peaks in the PK profile, as was the case here with both a fast absorption followed by a secondary delayed absorption.Many believe that the Linear Up Log Down method is the most accurate for interpolating between 2 adjacent datapoints because increase in concentration normally follow a linear path and decreases follow exponential decline. Thus the methodology for calculating AUC mirrors the expected profile path.
Where are the idosevar parameters set if you need to use them?
Idosevar gets created automatically by appending the word “Dose” to the name of the input column. For example, if your dosing input is “A1”, the corresponding idosevar is “A1Dose”. This is an internal variable name that can be used, in Secondary Parameter calculations. For example, if you wanted to calculate a dose-normalized Cmax using the above example, you could use the equation Cmax/A1Dose.
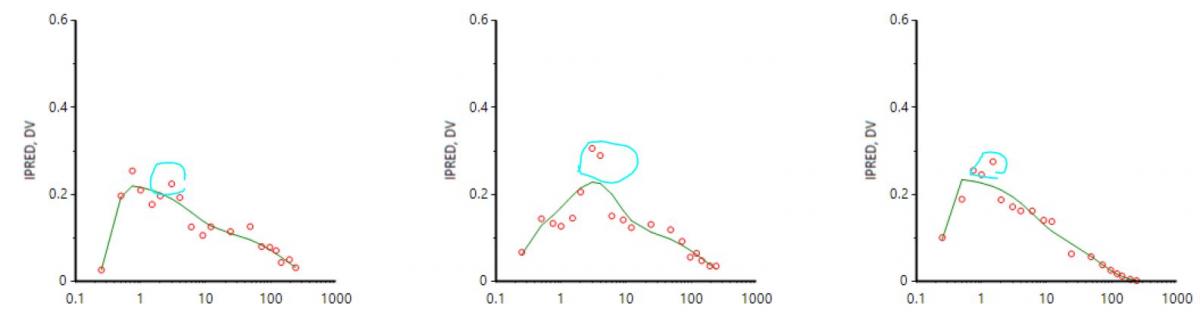
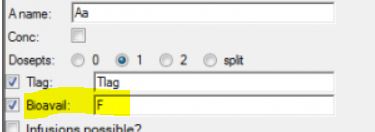
How to choose the initial value for Ka1 and Ka2?
One way to do this would be to calculate the slope of the log of the concentration values vs time. The slope of the first rise in log concentrations is an estimate of Ka1, and the slope of the second rise in log concentrations is an estimate of Ka2. These are crude estimates but generally good enough to achieve convergence. Another way is to use the Initial Estimates tab in the Phoenix model object. The second method was shown in the webinar. With all other parameters already having initial estimates via NCA methods, it was straightforward to find initial values for Ka1 and Ka2 using the overlay plot in the Initial Estimates tab.
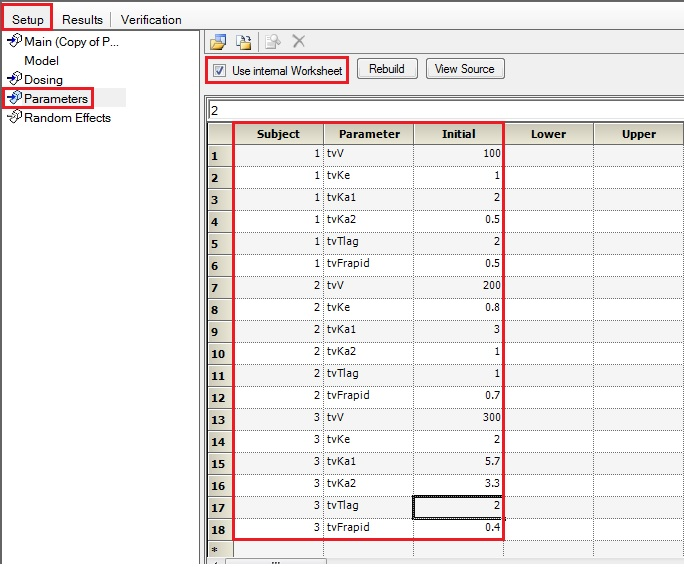
Can unique initial estimates for multiple individuals be input using the graphics mode?
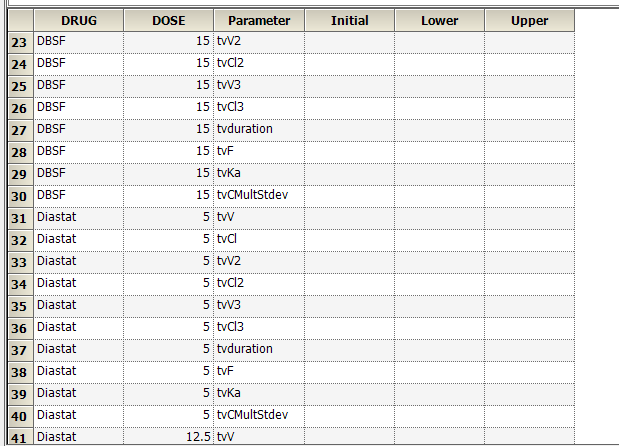
Yes. If you have multiple subjects in the input dataset and then use the Subject ID as a Sort variable in the model, you can go to Setup | Parameters, and then use an Internal Worksheet to specify initial parameter estimates for each Subject as shown below:
Why use micro parameterization?
There is no particular reason why the micro parameterization was selected here. The PML language supports micro, macro and clearance parameterizations. Generally the clearance parameterization is preferred as it is numerically more stable.
Is it possible to have covariate effects on Ka for multiple dose sites?
Yes, it is possible to have covariate effects on any of the structural parameters in a Phoenix model – ka1 and ka2 in this case. For example, you might have sub-populations that are “fast absorbers” and “slow absorbers” ka2.
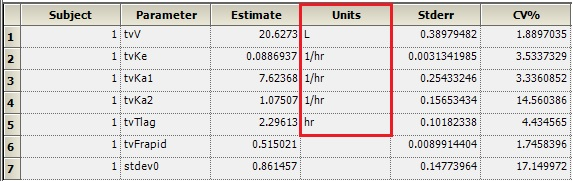
Is there a way to get the units to display for V or Ke like they do for Ka1, Ka2, and Tlag?
Dosing input units must be supplied to get units for V and Ke. The best way to do this is to create a Dosing input worksheet, with units in its column headers as shown below:

Then, map this worksheet into the dosing panel under Setup | Dosing. You will then get output units for parameters that depend on the Dose, such as V and Ke:
When and How to select lower and upper in initial estimates?
Typically one starts with unbounded parameters, with only initial estimates. Boundaries may be applied in a secondary run of the model if the Thetas are unexpected. For example, a Lower boundary might be 0 or 0.0001 to ensure that the final parameter remains positive. An upper boundary might be set to ensure that a final estimate falls below an expected maximum value for a volume or rate constant.Bounds are not recommended when using Phoenix models, and are not allowed for algorithms such as QRPEM. If your model generates unexpected values for Thetas, you may consider re-parameterizing your model or determining if you have an error in the PML. The use of bounds forces limits on certain parameters, but can mask problems in your model code that should be remedied.
When during the model development process do you estimate the standard error and care whether you have a var-covar matrix estimated versus when to be guided by the return code.
For individual model fitting (WNL mode) one selects a residual error model that best achieves normaility of the residuals (based on the residual plots). This is part of the basic model development process, along with identifying the best structural model. A variance–covariance matrix is automatically generated based on the fitted model.
The standard error is only needed for the final model to estimate the variability in the parameter estimates. One recommendation is to turn off standard errors during model building and then only calculate standard errors only on the final model.
Return codes do not reflect problems with the selection of the residual error model per se. The ideal return code is 1. If it is over than 1, that suggests that the program is having some difficulty achieving convergence. The actual fit may be okay, or it the program may have converged to a local minimum. If you do have a return code other than 1, we suggest you rerun the model with different initial estimates and check to see if minus 2 times the log-likelihood (-2*LL) stays the same (the initial fit is likely okay) or is reduced (indicative of a local minimum with the prior fit).
Edited by bwendt@certara.com, 21 November 2016 - 03:15 PM.